

Most American newborns will arrive home from the hospital and start hitting their developmental milestones, to their parents’ delight. They will hold their heads up by about three months. They will sit up by six. And they will walk around their first birthday. But about 1 in 10,000 will not. They will feel limp in their caregivers’ arms, won’t lift their heads, and will never learn to sit on their own. When their alarmed parents seek medical help, the babies will be diagnosed with spinal muscular atrophy, or SMA, a neuromuscular disease in which certain motor neurons of the spinal cord progressively deteriorate. The disease is triggered by a genetic malfunction that boils down to the gene called SMN2 (survival motor neuron 2), which causes bits of vital proteins to assemble incorrectly, resulting in progressive muscle weakness and paralysis.
Until five years ago, this diagnosis wasn’t far from a death sentence. SMA was considered the most common genetic cause of infant mortality. Many babies with SMA didn’t live to celebrate their second birthdays. Some lived past their toddlerhood, but never grew strong enough to run around or play with other kids and eventually succumbed to the disease. But in 2016 that dire prognosis changed for the first time in history—thanks to a new FDA-approved therapeutic developed by Adrian Krainer, a biochemist at Cold Spring Harbor Laboratory, in collaboration with Ionis Pharmaceuticals and Biogen.
Researchers think that RNA has huge untapped therapeutic potential.
Called Spinraza, the drug fixed the problem in a unique way. Administered through a spinal tap, Spinraza goes to work just as the SMN2’s garbled genetic code is transcribed into defective protein-making instructions—and corrects those instructions at the molecular level. Using more scientific terminology, Spinraza intervenes shortly after DNA is transcribed into RNA, a workhorse molecule responsible for many cellular processes, which in this case acts as a messenger carrying DNA’s instructions. “Spinraza is designed to bind to the messenger RNA, which enables the cell to handle it properly, and ultimately corrects the problem,” explains Krainer, who won a prestigious Wolf Prize in medicine this year, for his work explaining the molecular mechanisms behind this RNA process, which led to this new therapeutic.
Last year, messenger RNA, or mRNA, made the front pages of every newspaper as Pfizer and Moderna used the molecule to create COVID-19 vaccines. With this new method, never used before to vaccinate humans outside clinical trials, the drug makers employ mRNA to deliver specific instructions to our cells. The instructions tell the cells to generate the spike protein that coronavirus uses to infect us. Once produced inside the body, the spike protein draws the ire of the immune system, which remembers it as a foreign invader and is primed to fight the real coronavirus. After a while, the cells also destroy and remove any trace of the vaccine’s mRNA. The mRNA technology appeared novel, but it had been in the works, though out of the limelight, for years.
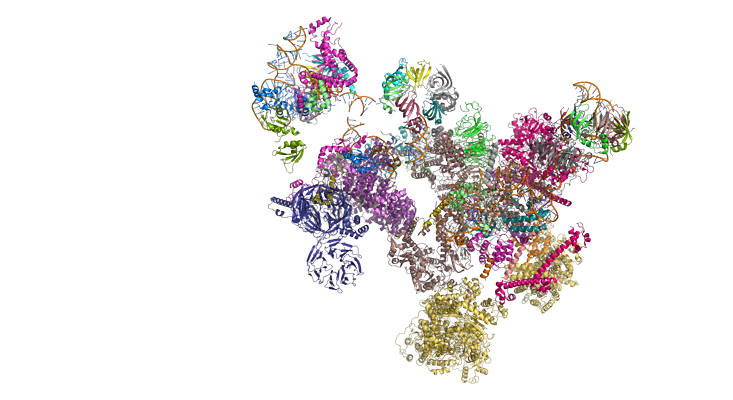
For decades, DNA has been scientists’ primary focus, while RNA was merely viewed as a helper, a passive carrier of genetic instructions, just an intermediary between DNA and proteins. “When it came to RNA, even scientists weren’t clear what was so important about it,” says Lynne Maquat who leads the Center for RNA Biology at the University of Rochester and who shared the Wolf Prize with Krainer. “People thought there were only three kinds of RNA, we already know what they do, end of story.”
But that view has changed. Not only did scientists discover many different types of RNAs, but they also realized that a huge portion of our DNA is devoted to making them. “We now know that at best only 3 percent of our genome codes for proteins,” says Joan Steitz, professor of molecular biophysics and biochemistry at Yale University, and another co-recipient of the Wolf Prize, who has been studying RNA since the 1960s. “And the other 97 percent is devoted to making all these different kinds of RNAs. We know what the most abundant and important of them do, but there are thousands of different ones that we still don’t have a full understanding of.” Building this understanding holds keys to treating many genetic disorders, which may originate in the faulty DNA, but can be corrected by mending the RNA or the processes in which this RNA is involved.
Researchers think that RNA has huge untapped therapeutic potential. Traditionally, when developing new meds, pharmaceutical companies target malfunctioning proteins that cause the disease. But targeting the RNA allows the problem to be corrected one step earlier, before the proteins are made, says Justin Kinney, a quantitative biologist at Cold Spring Harbor Laboratory who collaborates with Krainer to understand the inner workings of this molecule. “RNA is a great target for drug development,” Kinney says—because it is so versatile. His goal is to build a roadmap to a new generation of RNA-based therapeutics.
It’s not overly surprising that the DNA molecule overshadowed its less glamorous cousin for over half a century. After all, DNA’s charismatic double-helix string, tightly woven and nestled inside the cell nucleus, holds the code of life. Like a queen bee, the DNA molecule runs its cellular kingdom, dispatching orders for a myriad of cellular functions. Scientists who aimed to unveil the root-cause of genetic disease focused on the DNA.
But no queen can run her kingdom alone. A queen needs her messengers, maids, guards, and attachés. And that’s where the RNAs come in. Like their queen’s emissaries, RNA molecules carry out the instructions for protein assembly, catalyze reactions, and perform other duties, keeping their cellular dominion in good health.
Our understanding is so limited scientists sometimes don’t know why a drug works.
If you pictured each and every one of your cells as a bustling kingdom, you’d see a gazillion RNAs teeming around at all times. You would see the DNA being transcribed—its genetic instructions copied into messenger RNAs. These mRNAs would pass these instructions onto the ribosomes, the cellular protein and peptide-making machines, which would assemble them accordingly. To keep the conveyor going, transfer RNAs would deliver amino acids to this protein assembly line. And the specialized ribosomal RNAs would help stitch these amino acids into protein molecules. Meanwhile, more mRNAs are being forged—and just as they are produced, they are also getting spliced and diced for reasons scientists aren’t sure about. This is just one of the mysteries on which Kinney’s research might shed some light.
The act of transcribing DNA into mRNA begins when an enzyme called RNA polymerase binds to the DNA and starts copying the DNA sequence into an RNA sequence. But what comes out isn’t a very usable “draft.” For starters, the resulting mRNA is about 10 times longer than it should be, so it must be trimmed—or spliced, a process in which certain parts are kept and others are thrown out. This splicing is done by molecular machines called spliceosomes and involves removing the unnecessary nucleic acid sequences called introns (from “intervening” snippets) and stringing the remaining pieces, called exons, together.
“You can think of the RNA polymerase as a newspaper reporter and the spliceosomes as a very, very stringent editor that cuts 9 out of 10 paragraphs the reporter writes,” Kinney explains. “And it’s confusing why you would hire such a stringent editor to begin with—can’t your reporter just write less? So splicing seems like a very wasteful process. There are still debates about why it even evolved in the first place.”
The prevailing hypothesis is that it allows creating a greater variety of proteins, says Steitz, whose research elucidated the splicing mechanism—and human bodies need all of those proteins to function. Among other things, she found that the splicing process itself is governed by yet another RNA player—the tiny RNA-protein particles called snRNPs, or snurps. They find and remove these introns from the mRNA molecules.
In people with spinal muscular atrophy, this process hits a glitch. As introns are removed during splicing, one exon also gets axed from the resulting SMN2 RNA—exon 7. And without that exon, the proteins assembled with these RNA instructions come out defective, leading to spinal muscular atrophy.
Krainer likens the process to a cookbook with messed-up pages. “Our genome is like a library where thousands of books contain recipes for protein-making, with every chapter spelling out precise instructions, and in the right order,” he says. But in between the chapters there are extra pages (the introns) that shouldn’t be there. Splicing removes those pages, making reading straightforward. “If splicing is correct, you end up with perfect instructions. But in the case of SMN2, there’s a defect in Chapter 7, so splicing removes the entire chapter. Now a part of your instructions is missing, and you can’t follow the recipe.”
And that’s where Spinraza comes in, wielding its magic at the splicing level. The therapeutic is essentially a short piece of a DNA-like string, which binds to SMN2 RNA before that RNA is spliced. As it binds, it blocks various other proteins from messing up the splicing—and that allows exon 7 to be included. The resulting mRNA contains the correct protein-assembling instructions. The first approved drug of its kind, Spinraza is paving the way for other RNA-based therapies—and for good reasons.
RNA-based therapeutics can have a big advantage over the traditional protein-based ones. Currently, drug developers target malfunctioning proteins, aiming to fix their faults. But that’s a very intricate and failure-prone process, in which three things must come true, Kinney says. First, the drug must be able to bind to a spot, or a site, on the protein molecule. Second, it must correct the protein’s rogue behavior—for example, shut off the protein’s active site and disable its ability to cause harm. And lastly, it must not interfere with any other protein in the body, to avoid gumming up other vital functions.
“That’s a very difficult problem to solve,” Kinney says, because “most proteins don’t have a lot of potential binding targets.” RNA, on the contrary, is covered with binding sites because it is designed for other molecules to latch onto it. “The whole RNA is a target for drugs,” Kinney says. “The only limiting thing here is our understanding of how the RNA is controlled by various regulatory programs within the cell.”
“Our genome is like a library where thousands of books contain recipes for protein-making, with every chapter spelling out precise instructions.”
Our understanding is, in fact, so limited scientists sometimes don’t know why a drug works. Kinney cites one example. In 2020, the FDA approved another spinal muscular atrophy drug, Evrysdi. Compared to Spinraza, it’s a small molecule, only the size of about one base pair of DNA, and easier to make and administer—it can be taken orally. “It was essentially developed by trial and error,” Kinney says. “Scientists took a few hundred thousand random molecules and tested each one in cells to see which ones increase SMN2 exon 7 splicing. The initial candidate molecule then underwent years of testing and tweaking.” Although the final drug, Evrysdi, is safe and effective, scientists are still debating how it functions at the molecular level—in particular, how it singles out SMN2 exon 7 among the vast number of other exons.
Splicing is one of the most complex processes that occurs in human cells, and the spliceosome is the most complex piece of cellular machinery identified so far. In addition to the snRNPs, the spliceosome includes over a hundred different proteins. Incredibly, all of these interlocking molecular parts must assemble anew at each RNA intron. Decades of research unveiled how this complex machine operates once it assembles, but less is known about how the spliceosome recognizes the specific chunks of RNA it must cut out. “Understanding this requires new quantitative approaches,” Kinney explains.
That’s what Kinney is working on. Using high-precision experiments, mathematical modeling, and artificial intelligence, Kinney aims to clarify these mysteries at the level of molecular biophysics—how the spliceosome reads the RNA sequence and makes its cutting decisions, and how drugs like Evrysdi zero in on their specific targets. Splicing contributes to many diseases, including cystic fibrosis and cancer, and even those to which it doesn’t contribute, Kinney says, “may be treated by modulating the splicing process.” Once scientists elucidate the molecular cogwheels involved in splicing, they can determine where they get off track and correct them. “And that,” Kinney says, “will open a lot of opportunities for making new and better drugs.”
Lina Zeldovich grew up in a family of Russian scientists, listening to bedtime stories about volcanoes, black holes, and intrepid explorers. She has written for The New York Times, Scientific American, Reader’s Digest, and Audubon Magazine, among other publications, and won four awards for covering the science of poop. Her book, The Other Dark Matter: The Science and Business of Turning Waste into Wealth, will be released in October 2021 by Chicago University Press. You can find her at LinaZeldovich.com and @LinaZeldovich.
Support for this article was provided by Cold Spring Harbor Laboratory. Read more on the Nautilus channel, Biology + Beyond.
Lead image: CROCOTHERY / Shutterstock









