


In The Hitchhiker’s Guide to the Galaxy, Douglas Adams’ comedic sci-fi series from the 1970s, the Haggunenons of Vicissitus Three are one of the most insecure and angry life forms in the galaxy. What’s their problem? They have “impatient chromosomes” that instantly adapt to their surroundings. If they are sitting at a table, for instance, and are unable to reach a coffee spoon, “they are liable without a moment’s consideration to mutate into something with far longer arms … but which is probably quite incapable of drinking the coffee.”
Susan M. Rosenberg, a molecular biologist at Baylor College of Medicine, quotes Adams’ “(deliciously) askew” story in a research paper on mutations in evolution as an example of how, according to standard neo-Darwinian theory, evolution does not work. Organisms, all good students know, do not generate rapid genetic mutations in response to their environment. There are exceptions, such as mutations spurred by certain chemicals or radiation. In general, though, mutations, the raw material for natural selection, accumulate slowly in dividing cells, as the result of accidental errors in the way cells copy or repair their genetic material. The environment plays a role only later, selecting the most viable mutants.
But after more than two decades of experiments with the bacteria E. coli and most recently human cancer cells, Rosenberg is challenging that central tenet of evolutionary theory. According to Rosenberg and colleagues like Robert H. Austin, a physicist at Princeton University, organisms have evolved mechanisms that enable them to drive their own evolution in times of stress. Environmental pressure can boost mutation rates rapidly, even in cells that are not dividing, enabling them to adapt more quickly to new conditions. “Stop insisting that all mutations are random and evolution is slow is the entire story,” Austin says.

Contrary to traditional evolutionary theory, say Rosenberg and Austin—and they have their critics—cells might not need to divide much, or at all, to adapt. “All organisms’ genomes might be much more flexible and responsive than previously suspected,” Rosenberg has written. In practical terms, the findings suggest that scientists could upend the cellular mechanisms that allow bacteria to grow resistant to antibiotics. Medical experts could delay the development of resistance in cancer cells by making drugs that stop the stress response from turning on when cells are stressed by chemotherapies or other environmental conditions—approaches Rosenberg and Austin are already testing.
In 1943, Salvador Luria, an Italian-American microbiologist, and Max Delbrück, a German-American biologist, published an experiment that was part of the body of work for which they would be awarded the Nobel Prize. They let groups of identical bacteria grow for a while, and then infected each line with a phage (a virus that infects bacteria). After a while, they checked how many bacteria in each line survived the phage infection as a result of mutations that rendered them resistant to it.
If the mutations were caused by the stress from the phage infection, they reasoned, most of the mutations should appear around the time of the infection. The surviving phage-resistant bacterial colonies should have the same time to grow in the different lines and be similar in size.
But instead they found that the phage-resistant colonies in each line varied in size. That suggested that the stress didn’t cause the resistance mutations. Instead, the mutations must have been generated randomly in the bacteria’s genomes as the bacteria were dividing. This would have occurred at a different time for each line, explaining the size difference between the different lines of phage-resistant colonies.
Organisms under stress have a higher mutation rate, even though they are not dividing.
To be sure, this seminal experiment took place before DNA was identified as hereditary material. Still, it contributed to today’s view in evolutionary theory that mutations are largely generated randomly by internal processes in dividing cells, often as a result of errors in the way cells copy DNA. Because these errors are random, they’d be expected to be spread evenly across the genome.
But while the Luria-Delbrück experiment was solid science, it had a serious limitation, Austin and Rosenberg say: It didn’t leave any time for the bacteria to respond to the stress before the phages killed them. As a result, it couldn’t possibly detect any role of stress in causing mutations, a fact that Luria and Delbrück were aware of, Rosenberg says. “Everybody who didn’t already have a mutation that gave them phage resistance was dead,” she says.
However, in 1994, Rosenberg reported that E. coli bacteria under starvation stress used a different mechanism to generate mutations than when they weren’t starved. Because this suggested that stress might have something to do with mutations, she says, it was a big surprise, adding that many didn’t believe the results.
But that didn’t stop her. Rosenberg later found evidence that bacteria under starvation stress have a higher mutation rate, even though they are not dividing. She described one mechanism of this stress-induced “mutagenesis” in 2001: When stressed, the bacteria switched on polymerases (tiny protein-based machines, or enzymes, that copy DNA) that make more mistakes than the polymerases that copy DNA during cell division in unstressed cells. That’s because these “error-prone” polymerases lack the ability to replace incorrect base pairs (units of DNA) with correct ones in the newly copied DNA, or they add a wrong base pair or even skip a base pair.
At the time, researchers had just discovered that bacteria and humans had several error-prone polymerases, in addition to enzymes that are active during normal cell division. “Nobody knew what this particular subset or subfamily of error-prone polymerases were for,” Rosenberg says. “When we discovered a special DNA polymerase that came in just at that moment when they were stressed and produced mutations, that was like, OK, [cells] have a specialized mutation-generating machine.”
Rosenberg found that the mutations caused by these “error-prone” polymerases weren’t distributed evenly across the genome, but tended to appear more often in the areas next to where the polymerases were active. In 2012, she identified a network of 93 bacterial proteins that regulate whether stress responses turn on. Her discovery revealed that cells make mutations when they are poorly adapted to their environment—when they are stressed—and stress-induced mutations were neither generated randomly in time nor in terms of where they occurred in the genome. Rather, bacteria seemed to have evolved error-prone polymerases that enable them to drive their own evolution when stressed by boosting mutation rates. “Cells crank up genetic diversity and potentially increase their ability to evolve, specifically when they are poorly adapted to their environment,” Rosenberg says.
Researchers have since discovered stress-induced mutagenesis in other organisms, including in yeast, mouse, human cancer cells, and even plants and mice, though the mechanisms aren’t necessarily the same as what Rosenberg found in starving bacteria. “We started something that I think is now a bandwagon,” Rosenberg says.
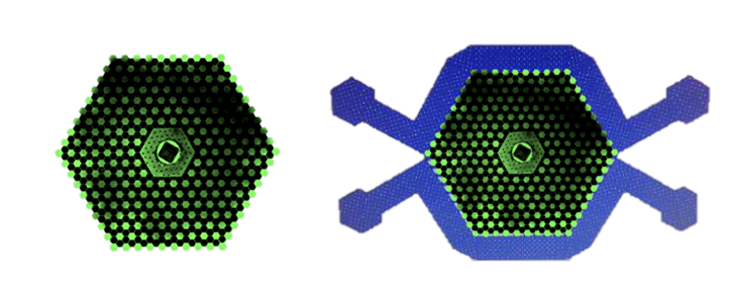
At Princeton, Austin and colleagues also exposed E. coli bacteria to stress—in this case an antibiotic—but with a twist. They wanted to see if the cells adapt to the stress more quickly in a complex environment, similar to the one bacteria typically encounter inside our bodies.
Austin says the idea for these experiments came from a 1932 paper by geneticist Sewall Wright. Wright proposed that a large population of organisms, where all members are connected and can interbreed, isn’t necessarily the best way for a beneficial adaptation to survive in the long run: There are too many competitors. A small, isolated population also isn’t ideal, because once the environment changes, maladapted organisms can’t leave, and well-adapted ones can’t come in.
Rather, Wright predicted, the best environment for beneficial adaptations to survive are small populations that are loosely connected, allowing the organisms to move and keep exchanging fresh genetic material, while still giving them a chance to establish themselves against the local competition. “It’s easier for the strongest guy to stand out and then propagate,” says Amy Wu, a former grad student in Austin’s lab.
To build such an environment for bacteria, Austin and colleagues made an inch-wide silicon chip with 1,200 tiny interconnected wells. Austin calls it the “death galaxy.” In 2011, Austin and colleagues reported that Wright’s prediction was correct. When Austin grew bacteria on the death galaxy chip while exposed to a range of different concentrations of the antibiotic Cipro (which doesn’t kill cells but keeps them from dividing) and nutrients, the cell’s progeny evolved resistant cells after just five hours. In contrast, no resistant cells appeared when the researchers grew bacteria across the same Cipro and nutrient concentration range on a flat, unstructured surface.
What’s more, the resistant bacteria that appeared on the death galaxy evolved the same four beneficial mutations in three independent experiments, including one that improved the ability of the bacteria to get rid of Cipro, and another that keeps Cipro from binding its target protein.
This evolutionary speed—the evolution of four beneficial mutations in five hours or at most 10 bacterial generations—is much faster than the time it takes bacteria to evolve beneficial mutations in other studies of bacterial evolution in a stress-free, homogeneous environment, Austin says. Experiments at Michigan State University, for example, found that it often takes hundreds of generations (several months) for a beneficial mutation to get established in an E. coli population grown in a homogeneous solution in flasks continuously mixed in a shaker.
“You may conclude that under harsh environmental conditions the mutation rate increases. But you could be mistaken.”
Austin’s death galaxy bacteria even evolved resistant cells—albeit more slowly—when he only added 100 cells initially. That’s too few cells, he says, for the mutations to have come from rare mutations that slowly accumulate in dividing cells—especially considering that the bacteria in the experiment didn’t divide much to begin with, because Cipro inhibits cell division. The resistance mutations, Austin says, must have been new, likely generated as a result of the same kind of stress-induced mutagenesis Rosenberg found in her experiments. Last year, Austin’s group reported that even single bacteria can evolve resistance to Cipro within a few hours. They did so without dividing, but after they multiplied their chromosomes a few times. Perhaps, Austin speculates, the chromosomes exchanged genetic material with each other.
A complex environment also seems to accelerate cancer cell evolution. When Wu grew such cells in the death galaxy environment across a range of different concentrations of a cancer drug, resistant cells evolved in about two weeks. That’s about 20 times faster than what would be expected if the mutations had come from spontaneous errors as dividing cells copy their DNA, again suggesting that stress-induced mutagenesis could be the cause.
The death galaxy experiments are important, Rosenberg says, because they simulate real-world conditions, and show that the structured, intricate environments in which pathogens and cells live inside our bodies affect rates of evolution profoundly. “Human bodies are not flasks of shaking homogeneous cultures,” she says; rather, they are quite heterogeneous, containing solid and liquid parts that likely harbor a range of different concentrations of chemicals and nutrients.
Austin has yet to show the exact mechanism of how mutations occur in his death galaxy experiments with cancer cells and bacteria. But to him, there’s no question that mechanisms like stress-induced mutagenesis are necessary to explain the pace of evolution in his experiments and in evolution in general. This is consistent, he says, with a famous calculation by J.B.S. Haldane, an evolutionary biologist. It suggests that if all mutations were slowly accelerating from random errors in dividing cells, it could take hundreds of generations to change one gene in a beneficial way. With thousands of genes in organisms like bacteria or humans, Austin says, “you just can’t explain the rate at which evolution has proceeded within the neo-Darwinist framework.”
Joanna Masel, an evolutionary biologist at the University of Arizona, says Austin’s death galaxy experiments provide one of the first direct experimental proofs she has seen that a complex environment with loosely connected populations can accelerate evolution.
But when it comes to modulation of mutation rates in response to environmental conditions, she and other evolutionary biologists are skeptical. First, it’s hard to show whether mutation rates are modulated by environmental conditions, says Paul Rainey, an evolutionary biologist at Massey University in New Zealand. That’s because it’s easy to miss some hidden growth period during which cells divide, in which case the mutations may still be created at the normal rate.
To be sure that a mutation is really a mutation, we need to capture it at the moment it occurred—and before any cell-division events take place. And even if cell division doesn’t occur, Rainey says, genes within cells can increase in copy number, similarly confounding attempts to obtain accurate measures of mutation rate. “If you don’t know what is happening within each cell, then you may conclude that under harsh environmental conditions the mutation rate increases,” Rainey says. “But you could be mistaken.”
It’s possible that stress-induced mutations play a role in the rapid evolution of the bacteria in Austin’s experiments, Masel says. However, this may mostly be due to the relatively small population size in the experiments. It may not be relevant for naturally occurring bacteria with their typically larger population size, she says, where mutation rate isn’t the limiting factor for evolutionary speed. “Large populations behave in a way that depends rather little on the mutation rate,” Masel says, because in large populations, a sufficient number of cells divide often enough to create enough mutations available for natural selection, even at the normal mutation rate.
Louise Johnson, an evolutionary biologist at the University of Reading in England, and her colleagues, recently demonstrated evolution can happen surprisingly fast due to selective pressures. When she removed two genes associated with movement in E. coli, the bacteria piled up in their culture dishes. As nutrients in their immediate environment ran out, that created a strong selective pressure. “There was suddenly a huge advantage of being able to move again,” Johnson says. Indeed, after 36 hours, some bacteria could move again because they had mutated another gene to become similar to one of the eliminated ones. The number of bacteria was in the billions, Johnson says, high enough to explain adaptation within the normal rate of mutations in a population.
What’s more, Haldane’s calculation that it could take hundreds of generations for one beneficial mutation to appear is likely too pessimistic, says Johannes Jaeger, an evolutionary systems biologist at the KLI research center near Vienna. He and others, including Andreas Wagner, an evolutionary biologist from the University of Zürich, have used computer simulations to show that biological systems might need far fewer genetic changes to adapt to a changing environment than Haldane assumed. That’s because biological systems are inherently “robust,” in that a phenotype remains stable and functioning even with a different organization at the molecular level. Jaeger recently showed that subtle differences in the collective activity of single genes that regulate each other can lead to changes in gene expressions that control fly development—and still lead to almost identical-looking flies.
Medical experts could delay the resistance in cancer cells to aggressive cancer drugs.
Even so, Jaeger and other evolutionary biologists agree that overall, most mutations are bad for an organism. And if most mutations are bad, why would a mechanism that accelerates mutation rates—like Rosenberg’s error-prone polymerases—evolve to make more mutations? Rainey thinks it wouldn’t. He explains the primary function of error-prone polymerases is to repair breaks in DNA. It’s therefore likely that any increase in the mutation rate is a consequence of that primary function, and not the cause of the error-prone polymerases’ evolution.
But Rosenberg counters that she and her colleagues found that stressed bacteria are not forced to use error-prone polymerase to repair damaged DNA; they use them even though it doesn’t necessarily lead to better repair. When researchers in her lab took starving bacteria and inactivated their major error-prone DNA polymerase, the stressed cells started to copy their DNA more accurately again. They seemed to have switched back to the more accurate DNA polymerase. This, however, didn’t seem to make any difference in the repair of damaged DNA. Stressed bacteria can in principle use the more accurate DNA polymerase, but use error-prone polymerases even though that does not give them an advantage in handling damaged DNA. This, Rosenberg says, suggests the possibility that use of these error-prone polymerases in DNA repair evolved because of their ability to cause more mutations—and not because they can better handle damaged DNA.
Rosenberg’s view recently got support from Lilach Hadany, a theoretical evolutionary biologist at Tel Aviv University. Hadany agrees with traditional evolutionary theory that most mutations are deleterious. But even so, mathematical models she developed show that bacteria can, in fact, evolve mechanisms like error-prone polymerases that increase mutagenesis in response to stress. “Stress-induced [mutagenesis], our model suggested, is not necessarily an accident,” she says. “It could actually happen, because it’s favored by selection.”
Interestingly, Masel says, this might be because most mutations are bad: Because increased mutation rates increase the number of less viable bacteria, more bacteria disappear from the population. This makes space for the more viable bacteria to spread, and enables the population to better survive in the long run. Hadany’s calculations convinced at least some evolutionary biologists, including Masel, to take the concept of mutation acceleration under stress more seriously. While there have been good reasons to be skeptical, Masel says, Hadany’s arguments “are proof the adaptive evolution of stress-induced mutagenesis could happen: Proving it has happened in any particular case is harder.”
Skepticism hasn’t kept Rosenberg and Austin from seeking medical applications of their findings. One application is cancer. Austin says his experiments suggest that putting too much stress on cancer cells by hitting them with high doses of cancer drugs could accelerate their evolution to develop drug resistance. “We give the patients as much as they can tolerate, guaranteeing the emergence of resistant cancer cells,” he says, adding that the current aggressive approach to cancer treatment has largely failed.
Instead, he is culturing cancer cells on his death galaxy to find the right low-dosing and timing of cancer drugs that keep the cancer cells from spreading without killing them—hopefully delaying the evolution of resistance as long as possible.
At least in an ovarian cancer model in mice, the approach seems to work. In 2009, Robert Gatenby, a radiologist at the Moffitt Cancer Center in Tampa, Florida, and his colleagues reported that interrupting, or down-adjusting, therapy as long as the tumor volume didn’t increase prolonged survival in these mice compared with the standard aggressive regimen. “If you give them standard high-dose therapy, the tumor can almost completely go away and then come back very rapidly and be resistant,” Gatenby says. “If you use an adaptive approach, we can consistently get control of the tumor.” Gatenby is now testing the approach in a 40-patient open clinical trial in patients with late-stage prostate cancer.
Rosenberg, meanwhile, wants to develop “anti-evolvability” drugs that inhibit some of the factors she found to regulate stress-induced mutation in bacteria. Such drugs might one day keep us from evolving resistance to antibiotics; perhaps they might even block the evolution of infectious pathogens themselves to allow the immune system to catch up. “My dream idea,” she says, “is maybe you don’t even need to give an antibiotic.” She adds that such drugs might be used as part of cancer therapies, and help keep cancer cells from becoming resistant to aggressive cancer drugs. “If that idea worked,” she says, “how wonderful that would be.”
Andreas von Bubnoff is a science writer based in Brooklyn.
Additional Reading
Al Mamun, A.A., et al. Identity and function of a large gene network underlying mutagenic repair of DNA breaks. Science 338, 1344-1348 (2012).
Bos, J., et al. Emergence of antibiotic resistance from multinucleated bacterial filaments. Proceedings of the National Academy of Sciences 112, 178-183 (2015).
Crombach, A., Wotton, K.R., Jimenez-Guri, E., & Jaeger, J. Gene Regulatory Dynamics Evolve along a Genotype Network. Molecular Biology and Evolution Advance Access (2016).
Gatenby, R.A., Silva, A.S., Gillies, R.J., & Frieden, B.R. Adaptive Therapy. Cancer Research 69, 4894 (2009).
Harris, R.S., Longerich, S., & Rosenberg, S.M. Recombination in adaptive mutation. Science 264, 258-260 (1994).
Luria, S.E. & Delbrück, M. Mutations of Bacteria from Virus Sensitivity to Virus Resistance. Genetics 28, 491-511 (1943).
McKenzie, G.J., Harris, R.S., Lee, P.L., & Rosenberg, S.M. The SOS response regulates adaptive mutation. Proceedings of the National Academy of Sciences 97, 6646-6651 (2000).
Ponder, R.A., Fonville, N.C., & Rosenberg, S.M. A switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation. Molecular Cell 19, 791-804 (2005).
Rosenberg, S.M. Evolving responsively: adaptive mutation. Nature Reviews Genetics 2, 504-515 (2001).
Rosenberg, S.M., Longerich, S., Gee, P., & Harris, R.S. Adaptive mutation by deletions in small mononucleotide repeats. Science 265, 405-407 (1994).
Shee, C., Gibson, J., Darrow, M., Gonzalez, C., & Rosenberg, S. Impact of a stress-inducible switch to mutagenic repair of DNA breaks on mutation in Escherichia coli. Proceedings of the National Academy of Sciences 108, 13659-13664 (2011).
Taylor, T., et al. Evolutionary resurrection of flagellar motility via rewiring of the nitrogen regulation system. Science 347, 1014-1017 (2015).
Wright, S. The roles of mutation, inbreeding, crossbreeding and selection in evolution. Proceedings of the sixth international congress of genetics, 356-366 (1932).
Zhang, Q., Robin, K., Liao, D., Lambert, G., & Austin, R. The Goldilocks Principle and Antibiotic Resistance in Bacteria. Molecular Pharmacology 8, 2063-2068 (2011).









